Reazioni di sostituzione del ligando. Argomento: Fasi elementari che coinvolgono la coordinazione e i composti organometallici nelle soluzioni e sulla superficie di metalli e ossidi. Meccanismo di sostituzione dei ligandi
I ligandi sono ioni o molecole direttamente associati all'agente complessante e sono donatori di coppie di elettroni. Questi sistemi ricchi di elettroni, con coppie di elettroni libere e mobili, possono essere donatori di elettroni, ad esempio: i composti di elementi p mostrano proprietà di formazione di complessi e agiscono come ligandi in un composto complesso. I ligandi possono essere atomi e molecole
(proteine, aminoacidi, acidi nucleici, carboidrati). L'efficienza e la forza dell'interazione donatore-accettore tra il ligando e l'agente complessante è determinata dalla loro polarizzabilità, ovvero la capacità della particella di trasformare i suoi gusci elettronici sotto l'influenza esterna.
Costante di fragilità:
Knest= 2 /
Alla bocca=1/Knest
Reazioni di sostituzione del ligando
Una delle fasi più importanti nella catalisi del complesso metallico, l'interazione del substrato Y con il complesso, avviene attraverso tre meccanismi:
a) Sostituzione del legante con un solvente. Questa fase è solitamente rappresentata come la dissociazione del complesso
L'essenza del processo nella maggior parte dei casi è la sostituzione del ligando con un solvente S, che viene poi facilmente sostituito da una molecola di substrato Y
b) Attaccamento di un nuovo ligando ad una coordinata libera con formazione di un associato seguita dalla dissociazione del ligando sostituito
c) Sostituzione sincrona (tipo S N 2) senza formazione intermedia
Idee sulla struttura dei metalloenzimi e di altri composti biocomplessi (emoglobina, citocromi, cobalamine). Principi fisico-chimici del trasporto dell'ossigeno da parte dell'emoglobina.
Caratteristiche della struttura dei metalloenzimi.
I composti biocomplessi variano significativamente in termini di stabilità. Il ruolo del metallo in tali complessi è altamente specifico: sostituirlo anche con un elemento simile nelle proprietà porta ad una perdita significativa o completa dell'attività fisiologica.
1. B12: contiene 4 anelli pirrolici, ione cobalto e gruppi CN. Promuove il trasferimento dell'atomo di H all'atomo di C in cambio di qualsiasi gruppo, partecipa al processo di formazione del desossiribosio dal ribosio.
2. emoglobina: ha una struttura quaternaria. Quattro catene polipeptidiche legate insieme formano una forma sferica quasi regolare, con ciascuna catena in contatto con due catene.
Emoglobina- un pigmento respiratorio che conferisce al sangue il suo colore rosso. L'emoglobina è costituita da proteine e porfirine di ferro e trasporta l'ossigeno dagli organi respiratori ai tessuti del corpo e da questi l'anidride carbonica agli organi respiratori.
Citocromi- proteine complesse (emoproteine) che effettuano il trasferimento graduale di elettroni e/o idrogeno dalle sostanze organiche ossidate all'ossigeno molecolare nelle cellule viventi. Questo produce il composto ricco di energia ATP.
Cobalamine- composti naturali di organocobalto biologicamente attivi. La base strutturale di K. è l'anello corrin, costituito da 4 nuclei pirrolici, in cui gli atomi di azoto sono legati all'atomo centrale di cobalto.
Principi fisico-chimici del trasporto dell'ossigeno da parte dell'emoglobina- L'atomo (Fe (II)) (uno dei componenti dell'emoglobina) è in grado di formare 6 legami di coordinazione. Di questi, quattro vengono utilizzati per fissare l'atomo Fe(II) stesso nell'eme, il quinto legame viene utilizzato per legare l'eme alla subunità proteica e il sesto legame viene utilizzato per legare la molecola di O2 o CO2.
Omeostasi del metallo-ligando e ragioni del suo disturbo. Il meccanismo dell'azione tossica dei metalli pesanti e dell'arsenico basato sulla teoria degli acidi e delle basi duri e morbidi (HSBA). Principi termodinamici della terapia chelante. Il meccanismo dell'azione citotossica dei composti del platino.
Nel corpo, si verifica continuamente la formazione e la distruzione di biocomplessi di cationi metallici e bioligandi (porfine, amminoacidi, proteine, polinucleotidi), che includono atomi donatori di ossigeno, azoto e zolfo. Lo scambio con l'ambiente mantiene le concentrazioni di queste sostanze a un livello costante, fornendo metallo ligando omeostasi. La violazione dell'equilibrio esistente porta a una serie di fenomeni patologici: stati di eccesso e carenza di metalli. Ad esempio, possiamo citare un elenco incompleto di malattie associate a cambiamenti nell'equilibrio metallo-legante per un solo ione: il catione rame. La carenza di questo elemento nel corpo provoca la sindrome di Menkes, la sindrome di Morphan, la malattia di Wilson-Konovalov, la cirrosi epatica, l'enfisema polmonare, l'aorto e l'arteriopatia, l'anemia. Un'assunzione eccessiva del catione può portare ad una serie di malattie di vari organi: reumatismi, asma bronchiale, infiammazioni dei reni e del fegato, infarto del miocardio, ecc., chiamate ipercupremia. È nota anche l'ipercupreosi professionale: la febbre del rame.
La circolazione dei metalli pesanti avviene in parte sotto forma di ioni o di complessi con aminoacidi e acidi grassi. Tuttavia, il ruolo principale nel trasporto dei metalli pesanti spetta alle proteine, che formano con essi forti legami.
Si fissano alle membrane cellulari e bloccano i gruppi tiolici delle proteine di membrana– Il 50% di essi sono proteine enzimatiche che interrompono la stabilità dei complessi proteici-lipidici della membrana cellulare e la sua permeabilità, provocando il rilascio di potassio dalla cellula e la penetrazione di sodio e acqua al suo interno.
Un tale effetto di questi veleni, che vengono fissati attivamente sui globuli rossi, porta alla rottura dell'integrità delle membrane degli eritrociti, all'inibizione dei processi di glicolisi aerobica e del metabolismo in essi in generale e all'accumulo di perossido di idrogeno emoliticamente attivo dovuto in particolare all'inibizione della perossidasi, che porta allo sviluppo di uno dei sintomi caratteristici dell'avvelenamento con composti di questo gruppo – l'emolisi.
La distribuzione e la deposizione di metalli pesanti e arsenico avviene in quasi tutti gli organi. Di particolare interesse è la capacità di queste sostanze di accumularsi nei reni, che si spiega con il ricco contenuto di gruppi tiolici nel tessuto renale, la presenza di una proteina in esso - metallobionina, contenente un gran numero di gruppi tiolici, che contribuisce alla deposizione a lungo termine di veleni. Anche il tessuto epatico, ricco di gruppi tiolici e contenente metallobionina, è caratterizzato da un elevato grado di accumulo di composti tossici di questo gruppo. Il periodo di deposito, ad esempio, del mercurio può raggiungere i 2 mesi o più.
Il rilascio di metalli pesanti e arsenico avviene in diverse proporzioni attraverso i reni, il fegato (con la bile), la mucosa dello stomaco e dell'intestino (con le feci), il sudore e le ghiandole salivari, i polmoni, che di solito è accompagnato da danni all'apparato escretore di questi organi e si manifesta con segni clinici corrispondenti.sintomi.
La dose letale per i composti solubili del mercurio è 0,5 g, per il calomelano 1–2 g, per il solfato di rame 10 g, per l'acetato di piombo 50 g, per la biacca 20 g, per l'arsenico 0,1–0,2 g.
La concentrazione di mercurio nel sangue è superiore a 10 μg/l (1γ%), nelle urine superiore a 100 μg/l (10γ%), la concentrazione di rame nel sangue è superiore a 1600 μg/l (160γ% ), l'arsenico è superiore a 250 μg/l (25γ).%) nelle urine.
La terapia chelante consiste nella rimozione di particelle tossiche
dal corpo, in base alla loro chelazione
complessonati di elementi s.
Farmaci utilizzati per l'eliminazione
sostanze tossiche incorporate nel corpo
le particelle sono chiamate disintossicanti.
Le reazioni dei composti di coordinazione avvengono sempre nella sfera di coordinazione di un metallo al quale sono legati dei ligandi. È quindi ovvio che affinché possa accadere qualcosa, i ligandi devono poter cadere in questa sfera. Ciò può avvenire in due modi:
- un complesso coordinativamente insaturo lega un nuovo ligando
- in una sfera di coordinazione già completata, un ligando viene sostituito da un altro.
Abbiamo già acquisito familiarità con il primo metodo quando abbiamo discusso dell'insaturazione di coordinazione e della regola dei 18 elettroni. Tratteremo qui il secondo.
Leganti di qualsiasi tipo possono essere sostituiti in qualsiasi combinazione
Ma di solito c'è una regola non detta: il numero di posti di coordinamento occupati non cambia. In altre parole, il conteggio degli elettroni non cambia durante la sostituzione. La sostituzione di un tipo di ligando con un altro è del tutto possibile e spesso avviene nella realtà. Prestiamo attenzione solo alla corretta gestione delle cariche quando si cambia il ligando L con il ligando X e viceversa. Se ci dimentichiamo di questo, lo stato di ossidazione del metallo cambierà e la sostituzione dei ligandi non è un processo di ossidoriduzione (se trovi o ti viene in mente un esempio contrario, fammi sapere: verrà automaticamente accreditato correttamente via, se non posso dimostrare che ti sei sbagliato, anche in questo caso ti garantisco un contributo positivo al karma).
Sostituzione che coinvolge ligandi apto
Con i ligandi più complessi non ci sono più difficoltà: basta ricordare una regola abbastanza ovvia: il numero di siti ligandi (cioè il numero totale di ligandi o centri ligandi di tipo X o L) viene mantenuto. Ciò deriva direttamente dalla conservazione del conteggio degli elettroni. Ecco alcuni esempi evidenti.

Prestiamo attenzione all'ultimo esempio. Il reagente di partenza per questa reazione è il dicloruro di ferro FeCl 2 . Fino a poco tempo fa avremmo detto: “È solo sale, cosa c’entra la chimica di coordinazione?” Ma non ci permetteremo più tale ignoranza. Nella chimica dei metalli di transizione non esistono “semplici sali”; tutti i derivati sono composti di coordinazione, ai quali valgono tutte le considerazioni sul conteggio degli elettroni, sulla configurazione d, sulla saturazione di coordinazione, ecc. Il cloruro di ferro, come siamo abituati a scriverlo, risulterebbe essere un complesso Fe(2+) di tipo MX 2 con configurazione d 6 e numero di elettroni 10. Non abbastanza! Bene? Dopotutto, abbiamo già capito che i ligandi possono essere impliciti. Per realizzare la reazione abbiamo bisogno di un solvente, e per tali reazioni molto probabilmente è THF. La dissoluzione del sale di ferro cristallino in THF avviene proprio perché il solvente donatore occupa spazi liberi, e l'energia di questo processo compensa la distruzione del reticolo cristallino. Non saremmo in grado di sciogliere questo “sale” in un solvente che non fornisce i servizi di solvatazione del metallo a causa della basicità di Lewis. In questo caso, e in un milione di casi simili, la solvatazione è semplicemente un’interazione di coordinazione. Scriviamo, solo per chiarezza, il risultato della solvatazione sotto forma del complesso FeX 2 L 4, in cui due ioni cloro rimangono nella sfera di coordinazione sotto forma di due leganti X, anche se molto probabilmente sono anche spostati da molecole del solvente donatore con formazione di un complesso carico FeL 6 2+. In questo caso non è così importante. In ogni caso, possiamo tranquillamente presumere di avere un complesso di 18 elettroni sia a sinistra che a destra.
La sostituzione, l'aggiunta e la dissociazione dei ligandi sono strettamente e inestricabilmente legate
Se ricordiamo la chimica organica, allora c'erano due meccanismi di sostituzione in un atomo di carbonio saturo: SN1 e SN2. Nella prima, la sostituzione avveniva in due fasi: prima il vecchio sostituente se ne andava, lasciando un orbitale vuoto sull'atomo di carbonio, che veniva poi occupato da un nuovo sostituente con una coppia di elettroni. Il secondo meccanismo presupponeva che l’uscita e l’arrivo avvenissero simultaneamente, di concerto, e che il processo fosse in una sola fase.
Nella chimica dei composti di coordinazione è del tutto possibile immaginare qualcosa di simile. Ma appare una terza possibilità, che l'atomo di carbonio saturo non aveva: prima attacchiamo un nuovo legante, poi stacchiamo quello vecchio. Diventa subito chiaro che questa terza opzione è difficilmente possibile se il complesso ha già 18 elettroni ed è saturo di coordinazione. Ma è del tutto possibile se il numero di elettroni è 16 o meno, cioè il complesso è insaturo. Ricordiamo immediatamente l'ovvia analogia con la chimica organica: anche la sostituzione nucleofila in un atomo di carbonio insaturo (in un anello aromatico o in un carbonio carbonilico) avviene prima come aggiunta di un nuovo nucleofilo e poi con l'eliminazione di quello vecchio.
Quindi, se abbiamo 18 elettroni, la sostituzione avviene come astrazione-addizione (gli appassionati di parole "intelligenti" usano il termine meccanismo dissociativo-associativo o semplicemente dissociativo). Un altro modo richiederebbe l’espansione della sfera di coordinazione fino a un conteggio di 20 elettroni. Questo non è assolutamente impossibile, e tali opzioni a volte vengono addirittura prese in considerazione, ma è sicuramente molto poco redditizio e ogni volta, in caso di sospetto di tale percorso, sono necessarie prove molto significative. Nella maggior parte di queste storie, i ricercatori alla fine conclusero di aver trascurato o tralasciato qualcosa e il meccanismo associativo fu rifiutato. Quindi, se il complesso originale ha 18 elettroni, prima deve andarsene un ligando, poi al suo posto deve prenderne uno nuovo, ad esempio:

Se vogliamo introdurre nella sfera di coordinazione un aptoligando che occupa più siti, dobbiamo prima liberarli tutti. Di norma, ciò avviene solo in condizioni abbastanza severe, ad esempio, per sostituire tre carbonili nel cromo carbonile con η 6 -benzene, la miscela viene riscaldata sotto pressione per molte ore, rilasciando di tanto in tanto il monossido di carbonio rilasciato. Sebbene nel diagramma si rappresenti la dissociazione di tre leganti con formazione di un complesso molto insaturo di 12 elettroni, in realtà la reazione molto probabilmente avviene per stadi, lasciando un carbonile alla volta, ed entrando nella sfera il benzene, aumentando gradualmente l'apticità, attraverso il stadi meno CO - digapto - meno un altro CO - tetraapto - meno un altro CO - esagapto, in modo che non si ottengano meno di 16 elettroni.

Quindi, se abbiamo un complesso con 16 elettroni o meno, allora la sostituzione del ligando molto probabilmente avviene come addizione-eliminazione (per chi ama le parole profonde: associativo-dissociativo o semplicemente associativo): il nuovo ligando viene prima , poi il vecchio se ne va. Sorgono due domande ovvie: perché il vecchio ligando se ne va, perché 18 elettroni sono molto buoni, e perché in questo caso non fare il contrario, come nei complessi di 18 elettroni. Alla prima domanda è facile rispondere: ogni metallo ha le sue abitudini, e alcuni metalli, soprattutto quelli tardivi, con i gusci D quasi completamente riempiti, preferiscono il conteggio di 16 elettroni e i corrispondenti tipi strutturali, e quindi buttano via il ligando extra , tornando alla loro configurazione preferita. A volte anche il fattore spaziale interferisce con la questione; i ligandi esistenti sono grandi e quello aggiunto sembra un passeggero di autobus nelle ore di punta. È più facile scendere e fare una passeggiata che soffrire in questo modo. Tuttavia, puoi spingere fuori un altro passeggero, lasciarlo fare una passeggiata e noi andremo. Anche la seconda domanda è semplice: in questo caso il meccanismo dissociativo dovrebbe prima fornire un complesso di 14 elettroni, e questo raramente è vantaggioso.
Ecco un esempio. Per varietà, sostituiamo il ligando X con un ligando L e non faremo confusione sugli stati di ossidazione e sulle cariche. Ancora una volta: durante la sostituzione, lo stato di ossidazione non cambia e, se il ligando X è scomparso, la perdita deve essere compensata dalla carica sul metallo. Se lo dimenticassimo, il numero di ossidazione diminuirebbe di 1, ma questo non è corretto.

E un'altra cosa strana. Si è formato un legame metallo-piridina a causa della coppia solitaria sull'azoto. In chimica organica, in questo caso mostreremmo sicuramente un vantaggio sull'azoto piridinico (ad esempio, in caso di protonazione o formazione di un sale quaternario), ma non lo facciamo mai in coordinazione chimica con la piridina o con qualsiasi altro L-ligando. Questo è terribilmente fastidioso per tutti coloro che sono abituati al sistema rigoroso e inequivocabile di disegnare strutture in chimica organica, ma dovrai abituarti, non è così difficile.
Ma non esiste un analogo esatto di SN2 nella chimica dei composti di coordinazione; ce n'è uno distante, ma è relativamente raro e non ne abbiamo realmente bisogno.
Leganti stabili e labili
Non potremmo assolutamente parlare dei meccanismi di sostituzione del ligando se non per una circostanza estremamente importante che utilizzeremo molto: la sostituzione del ligando, sia essa associativa o dissociativa, presuppone necessariamente la dissociazione del vecchio ligando. Ed è molto importante per noi sapere quali ligandi se ne vanno facilmente e quali se ne vanno male, preferendo rimanere nella sfera di coordinazione del metallo.
Come vedremo tra poco, in ogni reazione una parte dei ligandi rimane nella sfera di coordinazione e non cambia. Tali ligandi sono solitamente chiamati ligandi spettatori (se non vuoi parole così semplici e "non scientifiche", usa la parola inglese spectator nella trascrizione locale spectator, ligando-spettatore, ma, ti prego, non spettatore - questo è insopportabile! ). E alcuni partecipano direttamente alla reazione, trasformandosi in prodotti di reazione. Tali ligandi sono chiamati attori (non attori!), cioè attivi. È abbastanza chiaro che gli attori leganti devono essere facilmente introdotti e rimossi nella sfera di coordinazione del metallo, altrimenti la reazione semplicemente si bloccherà. Ma è meglio lasciare i ligandi spettatori nella sfera del coordinamento per molte ragioni, ma almeno per una cosa così banale come la necessità di evitare inutili storie intorno al metallo. È meglio che solo gli attori ligandi e nelle quantità richieste possano partecipare al processo desiderato. Se ci sono più siti di coordinazione disponibili del necessario, su di essi possono sedersi attori ligandi aggiuntivi e anche quelli che parteciperanno alle reazioni collaterali, riducendo la resa del prodotto target e la selettività. Inoltre, i ligandi spettatori svolgono quasi sempre molte funzioni importanti, ad esempio assicurano la solubilità dei complessi, stabilizzano il corretto stato di valenza del metallo, soprattutto se non è del tutto ordinario, aiutano i singoli stadi, forniscono stereoselettività, ecc. Non lo decifreremo ancora, perché discuteremo tutto questo in dettaglio quando arriveremo alle reazioni specifiche.
Si scopre che alcuni dei ligandi nella sfera di coordinazione dovrebbero essere strettamente legati e non inclini alla dissociazione e alla sostituzione con altri ligandi. Tali ligandi vengono solitamente chiamati coordinativamente stabile . O semplicemente stabile, se dal contesto è chiaro che stiamo parlando della forza dei legami dei ligandi, e non della loro stessa stabilità termodinamica, che non ci riguarda affatto.
E vengono chiamati ligandi che entrano ed escono facilmente e volentieri e sono sempre pronti a cedere il passo ad altri coordinazione labile , o semplicemente labile, e qui, per fortuna, non ci sono ambiguità.
Ciclobutadiene come ligando
Questo è probabilmente l'esempio più eclatante del fatto che nella sfera di coordinazione una molecola molto instabile può diventare un eccellente ligando e, per definizione, stabile nella coordinazione, se non altro perché se osa lasciare all'esterno la sfera calda e accogliente, niente di buono le farà attendetelo (a costo dell'output sarà proprio l'energia della destabilizzazione anti-aromatica).
Il ciclobutadiene e i suoi derivati sono gli esempi più noti di antiaromaticità. Queste molecole esistono solo a basse temperature e in una forma altamente distorta: per allontanarsi il più possibile dall'antiaromaticità, il ciclo viene distorto in un rettangolo allungato, eliminando la delocalizzazione e indebolendo al massimo la coniugazione dei doppi legami (questo è altrimenti chiamato l'effetto Jahn-Teller del 2° tipo: sistema degenere e il quadrato del ciclobutadiene è un biradicale degenere, ricordate il cerchio di Frost - è distorto e riduce la simmetria per rimuovere la degenerazione).
Ma nei complessi, il ciclobutadiene e i ciclobutadieni sostituiti sono eccellenti ligandi tetrahapto e la geometria di tali ligandi è esattamente un quadrato, con lunghezze di legame identiche. Come e perché ciò accada è una storia a parte, e non così ovvia come spesso si pensa.
Leganti labili di coordinazione
È necessario comprendere che non esiste una recinzione in cemento armato con filo spinato e torri di sicurezza tra le aree dei ligandi labili e stabili. Innanzitutto dipende dal metallo e LMKO funziona bene in questo contesto. Ad esempio, i metalli di transizione tardiva preferiscono i leganti morbidi, mentre i metalli di transizione precoce preferiscono quelli duri. Diciamo che lo ioduro si aggrappa molto strettamente agli atomi d 8 del palladio o del platino, ma raramente entra nella sfera di coordinazione del titanio o dello zirconio nella configurazione d 0. Ma in molti complessi metallici con caratteristiche meno pronunciate, lo ioduro si manifesta come un ligando completamente labile, cedendo facilmente il posto ad altri.
A parità di altre condizioni:
- I ligandi L sono generalmente più labili dei ligandi X;
- la labilità dei leganti X è determinata dalla durezza/morbidezza e dalla natura del metallo;
- I ligandi “impliciti” sono molto labili: solventi e ponti nei dimeri e nei cluster, tanto che la loro presenza nella sfera di coordinazione viene spesso del tutto trascurata e le strutture prive di essi vengono disegnate con una sfera di coordinazione formalmente insatura;
- I ligandi dihapto, ad esempio gli alcheni e gli alchini, si comportano come i tipici ligandi L: di solito sono abbastanza labili;
- i ligandi con maggiore apticità sono raramente labili, ma se un ligando poliapto può cambiare la modalità di legame in mono-apto, diventa più labile, ad esempio η 3 -allili si comportano in questo modo;
- i ligandi chelati che formano anelli chelati a 5 e 6 membri sono stabili, e i chelati con meno o più atomi dell'anello sono labili, almeno in un centro (l'anello chelato si apre e il ligando rimane sospeso come semplice). Ecco come si comporta l'acetato, ad esempio;
Leganti coordinativamente stabili
Ripetiamo tutto ancora, solo dall'altra parte
Nella sfera di coordinazione dei metalli, generalmente sono preservati (coordinazionalmente stabili):
- Chelanti a 5 e 6 membri;
- poliapto-ligandi: per eliminare i ciclopentadienili o il benzene (areni) dalla sfera di coordinazione, è necessario utilizzare tutti i tipi di tecniche speciali - semplicemente non escono, spesso resistono anche a un riscaldamento prolungato;
- ligandi legati a un metallo con un'elevata percentuale di effetto π-donatore (back-donazione);
- leganti molli per metalli di transizione tardiva;
- “ultimo” ligando nella sfera di coordinazione.
L'ultima condizione sembra strana, ma immagina un complesso che abbia molti ligandi diversi, tra i quali non ce ne sono assolutamente stabili (nessun chelante o poliaptoligando). Quindi, nelle reazioni, i ligandi cambieranno, relativamente parlando, in ordine di labilità relativa. I meno labili e gli ultimi a restare. Questo trucco si verifica, ad esempio, quando utilizziamo complessi di palladio fosfina. Le fosfine sono ligandi relativamente stabili, ma quando sono numerose e il metallo è ricco di elettroni (d 8, d 10), cedono il posto, una dopo l'altra, a ligandi attori. Ma l'ultimo legante della fosfina rimane solitamente nella sfera di coordinazione, e questo è molto positivo dal punto di vista delle reazioni a cui partecipano questi complessi. Torneremo più avanti su questa importante questione. Ecco un esempio abbastanza tipico in cui rimane solo una, “ultima” fosfina dalla sfera di coordinazione iniziale del complesso della fosfina di palladio nella reazione di Heck. Questo esempio ci avvicina molto al concetto più importante nelle reazioni dei complessi dei metalli di transizione: il concetto di controllo del ligando. Ne discuteremo più tardi.

Rimetallazione
Quando si sostituiscono alcuni ligandi con altri, è importante non esagerare con la reattività del ligando in entrata. Quando abbiamo a che fare con reazioni di molecole organiche, è importante per noi fornire esattamente una molecola di ciascun reagente nella sfera di coordinazione. Se entrano due molecole invece di una, c'è un'alta probabilità di reazioni collaterali che coinvolgono due ligandi identici. È possibile anche una perdita di reattività dovuta alla saturazione della sfera di coordinazione e all'impossibilità di introdurre in essa altri leganti necessari per il processo previsto. Questo problema si verifica particolarmente spesso quando nucleofili anionici forti, ad esempio carbanioni, vengono introdotti nella sfera di coordinazione. Per evitare ciò si utilizzano derivati meno reattivi, nei quali, al posto del catione di metallo alcalino, che determina l'elevata ionicità del legame, si utilizzano metalli e metalloidi meno elettropositivi (zinco, stagno, boro, silicio, ecc.), formando legami covalenti con la parte nucleofila. Le reazioni di tali derivati con derivati dei metalli di transizione producono prodotti di sostituzione del ligando, in linea di principio proprio come se il nucleofilo fosse in forma anionica, ma a causa della ridotta nucleofilicità con meno complicazioni e senza reazioni collaterali.
Tali reazioni di sostituzione del ligando sono solitamente chiamate transmetallazione per enfatizzare il fatto ovvio che il nucleofilo sembra cambiare i metalli: da più elettropositivi a meno elettropositivi. Questo nome, quindi, contiene un elemento di spiacevole schizofrenia: sembrava che avessimo già concordato di considerare tutte le reazioni dal punto di vista di un metallo di transizione, ma all'improvviso l'abbiamo perso di nuovo e abbiamo guardato questa e solo questa reazione. dal punto di vista di un nucleofilo. Dovrai essere paziente, è così che la terminologia si è sviluppata ed è accettata. In realtà, questa parola risale agli albori della chimica dei composti organometallici e al fatto che l'azione dei composti di litio o organomagnesio su alogenuri di vari metalli e metalloidi è uno dei metodi principali per la sintesi di tutti i composti organometallici, principalmente quelli di intransizione , e la reazione che stiamo ora considerando in chimica dei composti di coordinazione dei metalli di transizione è semplicemente una generalizzazione dell'antico metodo della chimica organometallica da cui tutto è nato.
![]()
Come avviene la transmetallazione?
La rimetallazione è simile alla sostituzione convenzionale e non simile. Sembra che se consideriamo un reagente organometallico di non transizione come semplicemente un carbanione con un controione, allora il legame del metallo di non transizione del carbonio è ionico. Ma questa idea sembra essere vera solo per i metalli più elettropositivi: il magnesio. Ma già per lo zinco e lo stagno questa idea è molto lontana dalla verità.
Pertanto, nella reazione entrano due legami σ e quattro atomi alle loro estremità. Di conseguenza, si formano due nuovi legami σ e quattro atomi si legano tra loro in un ordine diverso. Molto probabilmente, tutto ciò avviene simultaneamente in uno stato di transizione a quattro membri e la reazione stessa ha un carattere concertato, come tante altre reazioni dei metalli di transizione. L'abbondanza di elettroni e orbitali per letteralmente tutti i gusti e tutti i tipi di simmetrie rende i metalli di transizione capaci di mantenere contemporaneamente legami negli stati di transizione con diversi atomi.
Nel caso della rimetallazione, otteniamo un caso speciale di un processo molto generale, che viene chiamato semplicemente metatesi del legame σ. Non confonderli solo con la vera metatesi di olefine e acetileni, che sono reazioni catalitiche a tutti gli effetti con meccanismi propri. In questo caso stiamo parlando del meccanismo della transmetallazione o di un altro processo in cui avviene qualcosa di simile.

La principale reazione di sostituzione nelle soluzioni acquose, lo scambio di molecole d'acqua (22), è stata studiata per un gran numero di ioni metallici (Fig. 34). Lo scambio di molecole d'acqua nella sfera di coordinazione di uno ione metallico con la maggior parte delle molecole d'acqua presenti come solvente avviene molto rapidamente per la maggior parte dei metalli, e quindi la velocità di tale reazione potrebbe essere studiata principalmente con il metodo del rilassamento. Il metodo consiste nel disturbare l'equilibrio del sistema, ad esempio attraverso un forte aumento della temperatura. In nuove condizioni (temperatura più elevata), il sistema non sarà più in equilibrio. Viene quindi misurato il tasso di equilibrio. Se è possibile modificare la temperatura della soluzione all'interno 10-8 secondi, quindi puoi misurare la velocità di una reazione che richiede più di un periodo di tempo per essere completata 10-8 secondi.
È anche possibile misurare il tasso di sostituzione delle molecole d'acqua coordinate in vari ioni metallici con ligandi SO 2-4, S 2 O 3 2-, EDTA, ecc. (26). La velocità di questa reazione
dipende dalla concentrazione dello ione metallico idrato e non dipende dalla concentrazione del ligando in ingresso, il che rende possibile utilizzare l'equazione del primo ordine (27) per descrivere la velocità di questi sistemi. In molti casi, la velocità della reazione (27) per un dato ione metallico non dipende dalla natura del ligando in arrivo (L), che si tratti di molecole di H 2 O o di SO 4 2-, S 2 O 3 2-, o Ioni EDTA.
Questa osservazione, unita al fatto che l'equazione della velocità per questo processo non include la concentrazione del ligando influente, suggerisce che queste reazioni procedono secondo un meccanismo in cui la fase lenta comporta la rottura del legame tra lo ione metallico e l'acqua. Il composto risultante probabilmente coordina quindi rapidamente i ligandi vicini.
Nella sez. 4 di questo capitolo è stato affermato che gli ioni metallici idrati con carica più elevata, come Al 3+ e Sc 3+, scambiano le molecole d'acqua più lentamente degli ioni M 2+ e M +; Ciò fa supporre che la rottura dei legami svolga un ruolo importante nella fase che determina la velocità dell'intero processo. Le conclusioni ottenute in questi studi non sono conclusive, ma danno motivo di credere che i processi S N 1 siano importanti nelle reazioni di sostituzione degli ioni metallici idrati.
Probabilmente i composti complessi più studiati sono le ammine del cobalto (III). La loro stabilità, facilità di preparazione e reazioni lente li rendono particolarmente adatti per studi cinetici. Poiché gli studi su questi complessi sono stati condotti esclusivamente in soluzioni acquose, dovremmo prima considerare le reazioni di questi complessi con le molecole di solvente: l'acqua. Si è scoperto che in generale, le molecole di ammoniaca o ammina coordinate dallo ione Co(III) vengono sostituite così lentamente da molecole d'acqua che viene solitamente presa in considerazione la sostituzione di ligandi diversi dalle ammine.
È stata studiata la velocità delle reazioni del tipo (28) e si è scoperto che è del primo ordine rispetto al complesso del cobalto (X è uno dei tanti anioni possibili).
Poiché nelle soluzioni acquose la concentrazione di H 2 O è sempre approssimativa 55,5 M, allora è impossibile determinare l'effetto della modifica della concentrazione delle molecole d'acqua sulla velocità di reazione. Le equazioni di velocità (29) e (30) per una soluzione acquosa non sono distinguibili sperimentalmente, poiché k è semplicemente uguale a k" = k". Pertanto, è impossibile dire dall’equazione della velocità di reazione se l’H2O parteciperà alla fase che determina la velocità del processo. La risposta alla domanda se questa reazione procede mediante il meccanismo S N 2 con la sostituzione dello ione X con una molecola di H 2 O o mediante il meccanismo S N 1, che comporta prima la dissociazione seguita dall'aggiunta di una molecola di H 2 O, deve essere ottenuti utilizzando altri dati sperimentali.
Questo problema può essere risolto mediante due tipi di esperimenti. Tasso di idrolisi (sostituzione di uno ione Cl per molecola d'acqua) trance- + è circa 10 3 volte superiore alla velocità di idrolisi 2+. Un aumento della carica del complesso porta al rafforzamento dei legami metallo-ligando e, di conseguenza, all'inibizione della scissione di questi legami. Dovrebbero essere prese in considerazione anche l'attrazione dei ligandi in arrivo e la facilitazione della reazione di sostituzione. Poiché è stata riscontrata una diminuzione della velocità all'aumentare della carica del complesso, in questo caso sembra più probabile un processo dissociativo (S N 1).
Un altro metodo di prova si basa sullo studio dell'idrolisi di una serie di complessi simili trance-+. In questi complessi, la molecola di etilendiammina è sostituita da diammine simili, in cui gli atomi di idrogeno sull'atomo di carbonio sono sostituiti da gruppi CH 3. I complessi contenenti diammine sostituite reagiscono più velocemente del complesso etilendiamminico. La sostituzione degli atomi di idrogeno con gruppi CH 3 aumenta il volume del ligando, rendendo più difficile l'attacco dell'atomo di metallo da parte di un altro ligando. Questi ostacoli sterici rallentano la reazione attraverso il meccanismo S N 2. La presenza di ligandi voluminosi vicino all'atomo di metallo favorisce il processo dissociativo, poiché la rimozione di uno dei ligandi riduce il loro accumulo sull'atomo di metallo. L'aumento osservato nella velocità di idrolisi dei complessi con ligandi voluminosi è una buona prova che la reazione procede secondo il meccanismo S N 1.
Quindi, a seguito di numerosi studi sui complessi acidoamminici di Co(II), si è scoperto che la sostituzione dei gruppi acido con molecole d'acqua è un processo dissociativo in natura. Il legame atomo-ligando di cobalto viene esteso fino a un certo valore critico prima che le molecole d'acqua inizino a entrare nel complesso. Nei complessi con una carica pari o superiore a 2+, la rottura del legame cobalto-ligando è molto difficile e l'ingresso delle molecole d'acqua inizia a svolgere un ruolo più importante.
Si è riscontrato che la sostituzione del gruppo acido (X -) nel complesso cobalto(III) con un gruppo diverso dalla molecola H2O, (31) passa prima attraverso la sua sostituzione con una molecola
solvente - acqua, seguito dalla sua sostituzione con un nuovo gruppo Y (32).
Pertanto, in molte reazioni con complessi di cobalto(III), la velocità di reazione (31) è uguale alla velocità di idrolisi (28). Solo lo ione idrossile differisce dagli altri reagenti nella sua reattività con le ammine Co(III). Reagisce molto rapidamente con i complessi amminici del cobalto(III) (circa 10 6 volte più velocemente dell'acqua) a seconda del tipo di reazione idrolisi basica (33).
Questa reazione è risultata essere del primo ordine rispetto al legante sostitutivo OH - (34). Il secondo ordine complessivo della reazione e il progresso insolitamente rapido della reazione suggeriscono che lo ione OH - è un reagente nucleofilo eccezionalmente efficace per i complessi di Co(III) e che la reazione procede attraverso il meccanismo SN 2 attraverso la formazione di un intermedio.
Tuttavia, questa proprietà di OH - può essere spiegata anche con un altro meccanismo [equazioni (35), (36)]. Nella reazione (35), il complesso 2+ si comporta come un acido (secondo Brønsted), dando il complesso +, che è amido-(contenente)-composto - base corrispondente all'acido 2+.
La reazione procede quindi attraverso il meccanismo S N 1 (36) per formare un intermedio a cinque coordinate, che reagisce ulteriormente con le molecole di solvente per produrre il prodotto di reazione finale (37). Questo meccanismo di reazione è coerente con la velocità di una reazione del secondo ordine e corrisponde al meccanismo S N 1. Poiché la reazione nello stadio che determina la velocità coinvolge una base coniugata al complesso originale - l'acido, a questo meccanismo viene assegnata la designazione S N 1CB.
Determinare quale di questi meccanismi spiega meglio le osservazioni sperimentali è molto difficile. Tuttavia, ci sono prove convincenti a sostegno dell’ipotesi S N 1CB. I migliori argomenti a favore di questo meccanismo sono i seguenti: i complessi ottaedrici di Co(III) generalmente reagiscono attraverso il meccanismo dissociativo S N 1, e non esiste alcun argomento convincente sul perché lo ione OH - dovrebbe mediare il processo S N 2. È stato stabilito che lo ione ossidrile è un debole reagente nucleofilo nelle reazioni con Pt(II), e quindi la sua insolita reattività con Co(III) sembra irragionevole. Le reazioni con composti di cobalto(III) in mezzi non acquosi forniscono un'eccellente prova della formazione di intermedi a cinque coordinate forniti dal meccanismo S N 1 SV.
La prova finale è il fatto che in assenza di legami N - H nel complesso Co(III), reagisce lentamente con gli ioni OH -. Ciò, ovviamente, suggerisce che le proprietà acido-base del complesso sono più importanti delle proprietà nucleofile dell'OH per la velocità della reazione." Questa reazione dell'idrolisi basica dei complessi amminici Co (III) illustra il fatto che i dati cinetici spesso può essere interpretato in più di un modo, e per escludere l'uno o l'altro possibile meccanismo è necessario effettuare un esperimento piuttosto sottile.
Attualmente sono state studiate reazioni di sostituzione di un gran numero di composti ottaedrici. Se consideriamo i loro meccanismi di reazione, il più comune è il processo dissociativo. Questo risultato non è inaspettato poiché sei ligandi lasciano poco spazio attorno all'atomo centrale affinché altri gruppi possano attaccarsi ad esso. Ci sono solo pochi esempi in cui è stata dimostrata la presenza di un intermedio a sette coordinate o è stata rilevata l'influenza di un ligando intermedio. Pertanto, il meccanismo S N 2 non può essere completamente rifiutato come possibile via per le reazioni di sostituzione nei complessi ottaedrici.
Introduzione all'opera
Rilevanza dell'opera. I complessi di porfirine con metalli in stati di ossidazione elevati possono coordinare le basi in modo molto più efficiente dei complessi M 2+ e formare composti di coordinazione misti in cui nella prima sfera di coordinazione dell'atomo metallico centrale, insieme al ligando macrociclico, sono presenti acidi non ciclici ligandi e talvolta molecole coordinate. I problemi di compatibilità dei ligandi in tali complessi sono estremamente importanti, poiché è sotto forma di complessi misti che le porfirine svolgono le loro funzioni biologiche. Inoltre, le reazioni di addizione (trasferimento) reversibili di molecole di base, caratterizzate da costanti di equilibrio moderatamente elevate, possono essere utilizzate con successo per separare miscele di isomeri organici, per analisi quantitative e per scopi ambientali e medici. Pertanto, gli studi sulle caratteristiche quantitative e stechiometriche dell'equilibrio di coordinazione aggiuntiva sulle metalloporfirine (MP) e la sostituzione di ligandi semplici in esse sono utili non solo dal punto di vista della conoscenza teorica delle proprietà delle metalloporfirine come composti complessi, ma anche per risolvere il problema pratico della ricerca di recettori e trasportatori di piccole molecole o ioni. Ad oggi, sono praticamente assenti studi sistematici sui complessi di ioni metallici altamente carichi.
Obiettivo del lavoro. Questo lavoro è dedicato allo studio delle reazioni di complessi misti contenenti porfirine di cationi metallici altamente carichi Zr IV, Hf IV, Mo V e W V con basi N bioattive: imidazolo (Im), piridina (Py), pirazina (Pyz ), benzimidazolo (BzIm), stabilità della caratterizzazione e proprietà ottiche di complessi molecolari, dimostrazione dei meccanismi di reazione graduale.
Novità scientifica. Utilizzando i metodi di titolazione spettrofotometrica modificata, cinetica chimica, assorbimento elettronico e vibrazionale e spettroscopia 1 H NMR, sono state ottenute per la prima volta le caratteristiche termodinamiche e i meccanismi stechiometrici delle reazioni delle basi N con metalloporfirine con sfera di coordinazione mista (X) n -2 MTPP (X – acidoligando Cl - , OH) sono stati sostanziati - , O 2- , TPP - tetrafenilporfirina dianione). È stato stabilito che nella stragrande maggioranza dei casi, i processi di formazione delle supramolecole a base di metalloporfirina procedono gradualmente e comprendono diverse reazioni elementari reversibili e lente irreversibili di coordinazione delle molecole di base e sostituzione di ligandi acidi. Per ogni fase delle reazioni graduali, stechiometria, equilibrio o costanti di velocità, sono stati determinati gli ordini di reazioni lente basate sulla base e i prodotti sono stati caratterizzati spettralmente (spettri UV, visibili per i prodotti intermedi e UV, visibili e IR per i prodotti finali). Per la prima volta sono state ottenute equazioni di correlazione che consentono di prevedere la stabilità dei complessi supramolecolari con altre basi. Le equazioni vengono utilizzate nel lavoro per discutere il meccanismo dettagliato di sostituzione di OH - nei complessi Mo e W con una molecola di base. Vengono descritte le proprietà della MR, che la rendono promettente per l'uso nella rilevazione, separazione e analisi quantitativa di basi biologicamente attive, come stabilità moderatamente elevata dei complessi supramolecolari, una risposta ottica chiara e veloce, una soglia di sensibilità bassa e una seconda tempo di circolazione.
Significato pratico dell'opera. I risultati quantitativi e la fondatezza dei meccanismi stechiometrici delle reazioni di formazione di complessi molecolari sono di significativa importanza per la chimica di coordinazione dei ligandi macroeterociclici. Il lavoro di tesi mostra che i complessi misti contenenti porfirine mostrano un'elevata sensibilità e selettività verso le basi organiche bioattive, entro pochi secondi o minuti forniscono una risposta ottica adatta per il rilevamento pratico di reazioni con basi - COV, componenti di farmaci e prodotti alimentari, a causa a cui è consigliato l'uso come componenti di sensori di base in ecologia, industria alimentare, medicina e agricoltura.
Approvazione del lavoro. I risultati del lavoro sono stati riportati e discussi in:
IX Conferenza Internazionale sui Problemi di Solvazione e Complessazione nelle Soluzioni, Ples, 2004; XII Simposio sulle interazioni intermolecolari e conformazioni delle molecole, Pushchino, 2004; XXV, XXVI e XXIX sessioni scientifiche del seminario russo sulla chimica delle porfirine e dei loro analoghi, Ivanovo, 2004 e 2006; VI Conferenza scolastica di giovani scienziati dei paesi della CSI sulla chimica delle porfirine e dei composti correlati, San Pietroburgo, 2005; VIII Scuola Scientifica - Conferenza di Chimica Organica, Kazan, 2005; Conferenza scientifica tutta russa “Composti macrociclici naturali e loro analoghi sintetici”, Syktyvkar, 2007; XVI Conferenza Internazionale sulla Termodinamica Chimica in Russia, Suzdal, 2007; XXIII Conferenza Internazionale Chugaev sulla Chimica di Coordinazione, Odessa, 2007; Conferenza internazionale su porfirine e ftalocianine ISPP-5, 2008; 38a Conferenza Internazionale sulla Chimica di Coordinazione, Israele, 2008.
Capitolo 17. Connessioni complesse
17.1. Definizioni di base
In questo capitolo acquisirai familiarità con un gruppo speciale di sostanze complesse chiamate completo(O coordinazione) connessioni.
Attualmente, una definizione rigorosa del concetto " particella complessa" NO. Solitamente viene utilizzata la seguente definizione.
Ad esempio, uno ione rame idrato 2 è una particella complessa, poiché esiste effettivamente in soluzioni e in alcuni idrati cristallini, è formato da ioni Cu 2 e molecole di H 2 O, le molecole d'acqua sono molecole reali e gli ioni Cu 2 esistono nei cristalli di molti composti del rame. Al contrario, lo ione SO 4 2 non è una particella complessa, poiché, sebbene gli ioni O 2 siano presenti nei cristalli, lo ione S 6 non esiste nei sistemi chimici.
Esempi di altre particelle complesse: 2, 3, , 2.
Allo stesso tempo, gli ioni NH 4 e H 3 O sono classificati come particelle complesse, sebbene gli ioni H non esistano nei sistemi chimici.
A volte le particelle chimiche complesse sono chiamate particelle complesse, tutti o parte dei legami in cui si formano secondo il meccanismo donatore-accettore. Nella maggior parte delle particelle complesse questo è il caso, ma, ad esempio, nell'allume di potassio SO 4 nella particella complessa 3, il legame tra gli atomi di Al e O è effettivamente formato secondo il meccanismo donatore-accettore, e nella particella complessa c'è solo un'interazione elettrostatica (ione-dipolo). Ciò è confermato dall'esistenza nell'allume ferro-ammonio di una particella complessa simile nella struttura, in cui è possibile solo l'interazione ione-dipolo tra le molecole d'acqua e lo ione NH 4.
In base alla loro carica, le particelle complesse possono essere cationi, anioni o molecole neutre. I composti complessi contenenti tali particelle possono appartenere a diverse classi di sostanze chimiche (acidi, basi, sali). Esempi: (H 3 O) è un acido, OH è una base, NH 4 Cl e K 3 sono sali.
Tipicamente l'agente complessante è un atomo dell'elemento che forma il metallo, ma può anche essere un atomo di ossigeno, azoto, zolfo, iodio e altri elementi che formano non metalli. Lo stato di ossidazione dell'agente complessante può essere positivo, negativo o nullo; quando un composto complesso è formato da sostanze più semplici, non cambia.
I ligandi possono essere particelle che, prima della formazione di un composto complesso, erano molecole (H 2 O, CO, NH 3, ecc.), anioni (OH, Cl, PO 4 3, ecc.), nonché un catione idrogeno . Distinguere non identificato o ligandi monodentati (collegati all'atomo centrale tramite uno dei suoi atomi, cioè tramite un legame), bidentato(collegato all'atomo centrale tramite due dei suoi atomi, cioè tramite due legami), tridentato eccetera.
Se i ligandi non sono identificati, il numero di coordinazione è uguale al numero di tali ligandi.
Il CN dipende dalla struttura elettronica dell'atomo centrale, dal suo stato di ossidazione, dalla dimensione dell'atomo centrale e dei ligandi, dalle condizioni per la formazione del composto complesso, dalla temperatura e da altri fattori. CN può assumere valori da 2 a 12. Molto spesso è sei, un po' meno spesso quattro.
Esistono particelle complesse con diversi atomi centrali.
Vengono utilizzati due tipi di formule strutturali di particelle complesse: che indicano la carica formale dell'atomo centrale e dei ligandi, o che indicano la carica formale dell'intera particella complessa. Esempi:

Per caratterizzare la forma di una particella complessa, viene utilizzato il concetto di poliedro di coordinazione (poliedro).

I poliedri di coordinazione includono anche un quadrato (CN = 4), un triangolo (CN = 3) e un manubrio (CN = 2), sebbene queste figure non siano poliedri. Esempi di poliedri di coordinazione e particelle complesse con forme corrispondenti per i valori CN più comuni sono mostrati in Fig. 1.
17.2. Classificazione dei composti complessi
Come sostanze chimiche, i composti complessi sono divisi in composti ionici (a volte vengono chiamati ionico) e molecolare ( non ionico) connessioni. I composti ionici complessi contengono particelle complesse cariche - ioni - e sono acidi, basi o sali (vedere § 1). I composti complessi molecolari sono costituiti da particelle complesse non cariche (molecole), ad esempio: oppure - classificarli in qualsiasi classe principale di sostanze chimiche è difficile.
Le particelle complesse incluse nei composti complessi sono piuttosto diverse. Pertanto, per classificarli vengono utilizzate diverse caratteristiche di classificazione: il numero di atomi centrali, il tipo di ligando, il numero di coordinazione e altri.
Secondo il numero di atomi centrali le particelle complesse sono suddivise in single core E multicore. Gli atomi centrali delle particelle complesse multinucleari possono essere collegati tra loro direttamente o tramite ligandi. In entrambi i casi gli atomi centrali con i ligandi formano un'unica sfera interna del composto complesso:


In base al tipo di ligandi, le particelle complesse vengono suddivise in
1) Complessi acquatici, cioè particelle complesse in cui le molecole d'acqua sono presenti come ligandi. Gli acquacomplessi cationici m sono più o meno stabili, gli acquacomplessi anionici sono instabili. Tutti gli idrati di cristallo appartengono a composti contenenti acquacomplessi, ad esempio:
Mg(ClO4) 2. 6H 2 O è in realtà (ClO 4) 2;
BeSO4. 4H 2 O è in realtà SO 4;
Zn(BrO3) 2. 6H 2 O è in realtà (BrO 3) 2;
CuSO4. 5H 2 O è in realtà SO 4. H2O.
2) Complessi idrossilici, cioè particelle complesse in cui sono presenti come ligandi gruppi idrossilici, che erano ioni idrossido prima di entrare nella composizione della particella complessa, ad esempio: 2, 3, .
I complessi idrossilici sono formati da complessi acquosi che presentano le proprietà degli acidi cationici:
2+4OH = 2+4H2O
3) Ammoniaca, cioè particelle complesse in cui i gruppi NH 3 sono presenti come ligandi (prima della formazione di una particella complessa - molecole di ammoniaca), ad esempio: 2, , 3.
L'ammoniaca può anche essere ottenuta da complessi acquatici, ad esempio:
2+4NH3 = 2+4H2O
Il colore della soluzione in questo caso cambia dal blu al blu oltremare.
4) Complessi acidi, cioè particelle complesse in cui residui acidi di acidi sia privi di ossigeno che contenenti ossigeno sono presenti come ligandi (prima della formazione di una particella complessa - anioni, ad esempio: Cl, Br, I, CN, S 2, NO 2, S 2 O 3 2 , CO 3 2 , C 2 O 4 2 , ecc.).
Esempi di formazione di complessi acidi:
Hg2 + 4I = 2
AgBr + 2S 2 O 3 2 = 3 + Br
Quest'ultima reazione viene utilizzata in fotografia per rimuovere il bromuro d'argento non reagito dai materiali fotografici.
(Quando si sviluppa pellicola fotografica e carta fotografica, la parte non esposta del bromuro d'argento contenuto nell'emulsione fotografica non viene ridotta dallo sviluppatore. Per rimuoverla si utilizza questa reazione (il processo è chiamato “fissaggio”, poiché il bromuro d'argento non rimosso si decompone gradualmente alla luce, distruggendo l'immagine)
5) I complessi in cui gli atomi di idrogeno sono i ligandi si dividono in due gruppi completamente diversi: idruro complessi e complessi inclusi nella composizione onio connessioni.
Durante la formazione dei complessi idruro – , , – l'atomo centrale è un accettore di elettroni e il donatore è lo ione idruro. Lo stato di ossidazione degli atomi di idrogeno in questi complessi è –1.
Nei complessi di onio, l'atomo centrale è un donatore di elettroni e l'accettore è un atomo di idrogeno nello stato di ossidazione +1. Esempi: H 3 O o – ione ossonio, NH 4 o – ione ammonio. Inoltre, esistono derivati sostituiti di tali ioni: – ione tetrametilammonio, – ione tetrafenilarsonio, – ione dietilossonio, ecc.
6) Carbonile complessi - complessi in cui i gruppi CO sono presenti come ligandi (prima della formazione del complesso - molecole di monossido di carbonio), ad esempio: , , ecc.
7) Alogenati anionici complessi – complessi di tipo .
In base alla tipologia dei ligandi si distinguono anche altre classi di particelle complesse. Inoltre, esistono particelle complesse con diversi tipi di ligandi; L'esempio più semplice è il complesso acqua-idrossi.
17.3. Nozioni di base sulla nomenclatura dei composti complessi
La formula di un composto complesso è compilata allo stesso modo della formula di qualsiasi sostanza ionica: in primo luogo è scritta la formula del catione e in secondo luogo quella dell'anione.
La formula di una particella complessa è scritta tra parentesi quadre nella seguente sequenza: viene posto prima il simbolo dell'elemento che forma il complesso, poi le formule dei ligandi che erano cationi prima della formazione del complesso, poi le formule dei ligandi che erano molecole neutre prima della formazione del complesso, e dopo di esse le formule dei ligandi, che erano anioni prima della formazione del complesso.
Il nome di un composto complesso è costruito allo stesso modo del nome di qualsiasi sale o base (gli acidi complessi sono chiamati sali di idrogeno o ossonio). Il nome del composto include il nome del catione e il nome dell'anione.
Il nome della particella complessa include il nome dell'agente complessante e i nomi dei ligandi (il nome è scritto secondo la formula, ma da destra a sinistra. Per gli agenti complessanti, i nomi russi degli elementi sono usati nei cationi e quelli latini negli anioni.
Nomi dei ligandi più comuni:
| H2O – acqua | Cl – cloro | SO 4 2 – solfato | OH – idrossi |
| CO – carbonile | Br – bromo | CO 3 2 – carbonato | H – idrato |
| NH 3 – ammina | NO2 – nitro | CN – ciano | NO – nitroso |
| NO – nitrosile | O2 – osso | NCS – tiocianato | H+I – idro |
Esempi di nomi di cationi complessi:
Esempi di nomi di anioni complessi:
2 – ione tetraidrossizincato
3 – ione di(tiosolfato)argentato(I).
3 - ione esacianocromato (III).
– ione tetraidrossidiaquaalluminato
– ione tetranitrodiammina cobaltato(III).
3 - ione pentacianoaquaferrato (II).
Esempi di nomi di particelle complesse neutre:
Regole di nomenclatura più dettagliate sono fornite nei libri di consultazione e nei manuali speciali.
17.4. Legami chimici nei composti complessi e loro struttura
Nei composti complessi cristallini con complessi carichi, il legame tra il complesso e gli ioni della sfera esterna è ionico, i legami tra le restanti particelle della sfera esterna sono intermolecolari (incluso l'idrogeno). Nei composti complessi molecolari, la connessione tra i complessi è intermolecolare.
Nella maggior parte delle particelle complesse, i legami tra l'atomo centrale e i ligandi sono covalenti. Tutti o parte di essi si formano secondo il meccanismo donatore-accettante (di conseguenza - con un cambiamento negli oneri formali). Nei complessi meno stabili (ad esempio, negli acquacomplessi di elementi alcalini e alcalino-terrosi, nonché di ammonio), i ligandi sono trattenuti per attrazione elettrostatica. Il legame nelle particelle complesse è spesso chiamato legame donatore-accettore o legame di coordinazione.
Consideriamo la sua formazione usando l'esempio dell'acquacazione del ferro (II). Questo ione si forma dalla reazione:
FeCl2cr + 6H2O = 2 + 2Cl
La formula elettronica dell'atomo di ferro è 1 S 2 2S 2 2P 6 3S 2 3P 6 4S 2 3D 6. Disegniamo un diagramma dei sottolivelli di valenza di questo atomo:
Quando si forma uno ione con doppia carica, l'atomo di ferro perde due 4 S-elettrone:
Lo ione ferro accetta sei coppie di elettroni di atomi di ossigeno di sei molecole d'acqua negli orbitali di valenza liberi:

Si forma un catione complesso la cui struttura chimica può essere espressa con una delle seguenti formule:

La struttura spaziale di questa particella è espressa da una delle formule spaziali:

La forma del poliedro di coordinazione è ottaedro. Tutti i legami Fe-O sono uguali. Ipotetico sp 3 D 2 - Ibridazione AO dell'atomo di ferro. Le proprietà magnetiche del complesso indicano la presenza di elettroni spaiati.
Se FeCl 2 viene sciolto in una soluzione contenente ioni cianuro, si verifica la reazione
FeCl 2cr + 6CN = 4 + 2Cl.
Lo stesso complesso si ottiene aggiungendo una soluzione di cianuro di potassio KCN ad una soluzione di FeCl 2:
2 + 6CN = 4 + 6H 2 O.
Ciò suggerisce che il complesso del cianuro è più forte del complesso dell'acqua. Inoltre, le proprietà magnetiche del complesso del cianuro indicano l'assenza di elettroni spaiati nell'atomo di ferro. Tutto ciò è dovuto alla struttura elettronica leggermente diversa di questo complesso:

I ligandi CN “più forti” formano legami più forti con l’atomo di ferro, il guadagno di energia è sufficiente per “infrangere” la regola di Hund e rilasciare 3 D-orbitali per coppie solitarie di ligandi. La struttura spaziale del complesso del cianuro è la stessa dell'acquacomplesso, ma il tipo di ibridazione è diverso: D 2 sp 3 .
La “forza” del ligando dipende principalmente dalla densità elettronica della nuvola di coppie solitarie di elettroni, cioè aumenta al diminuire della dimensione atomica, al diminuire del numero quantico principale, dipende dal tipo di ibridazione EO e da alcuni altri fattori . I ligandi più importanti possono essere disposti in una serie di “forza” crescente (una sorta di “serie di attività” dei ligandi), questa serie è chiamata serie spettrochimiche di ligandi:
| IO; Fratello; : SCN, CI, F, OH, H2O; : NCS, NH3; SO3S : 2 ; : CN, CO |
Per i complessi 3 e 3 gli schemi formativi sono i seguenti:
|
|

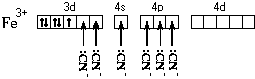
Per complessi con CN = 4 sono possibili due strutture: tetraedrico (nel caso sp 3-ibridazione), ad esempio 2, e un quadrato piatto (nel caso sp 2-ibridazione), ad esempio, 2.
17.5. Proprietà chimiche dei composti complessi
I composti complessi sono caratterizzati principalmente dalle stesse proprietà dei composti ordinari delle stesse classi (sali, acidi, basi).
Se il composto complesso è un acido allora è un acido forte; se è una base allora è una base forte. Queste proprietà dei composti complessi sono determinate solo dalla presenza di ioni H 3 O o OH. Inoltre, acidi complessi, basi e sali entrano nelle normali reazioni di scambio, ad esempio:
SO4 + BaCl2 = BaSO4 + Cl2
FeCl3 + K4 = Fe43 + 3KCl
L'ultima di queste reazioni viene utilizzata come reazione qualitativa per gli ioni Fe 3. La sostanza insolubile risultante di colore oltremare è chiamata “blu di Prussia” [nome sistematico: esacianoferrato di ferro (III)-potassio (II).
Inoltre, la particella complessa stessa può entrare in una reazione e più è attiva, meno è stabile. Tipicamente si tratta di reazioni di sostituzione del ligando che si verificano in soluzione, ad esempio:
2 + 4NH3 = 2 + 4H2O,
così come reazioni acido-base come
2+2H3O = +2H2O
2+2OH = +2H2O
Il prodotto formato in queste reazioni, dopo isolamento ed essiccazione, si trasforma in idrossido di zinco:
Zn(OH)2 + 2H2O
L'ultima reazione è l'esempio più semplice della decomposizione di un composto complesso. In questo caso avviene a temperatura ambiente. Altri composti complessi si decompongono se riscaldati, ad esempio:
SO4. H 2 O = CuSO 4 + 4NH 3 + H 2 O (sopra 300 o C)
4K 3 = 12KNO 2 + 4CoO + 4NO + 8NO 2 (sopra 200 o C)
K 2 = K 2 ZnO 2 + 2H 2 O (sopra 100 o C)
Per valutare la possibilità di una reazione di sostituzione del ligando, è possibile utilizzare una serie spettrochimica, guidata dal fatto che i ligandi più forti spostano quelli meno forti dalla sfera interna.
17.6. Isomeria di composti complessi
È associato l'isomeria dei composti complessi
1) con possibili diverse disposizioni di ligandi e particelle della sfera esterna,
2) con una diversa struttura della particella complessa stessa.
Il primo gruppo comprende idrato(generalmente solvatare) E ionizzazione isomerismo, al secondo - spaziale E ottico.
L'isomeria dell'idrato è associata alla possibilità di una diversa distribuzione delle molecole d'acqua nelle sfere esterna ed interna di un composto complesso, ad esempio: (colore rosso-marrone) e Br 2 (colore blu).
L'isomeria di ionizzazione è associata alla possibilità di diverse distribuzioni di ioni nelle sfere esterna ed interna, ad esempio: SO 4 (viola) e Br (rosso). Il primo di questi composti forma un precipitato reagendo con una soluzione di cloruro di bario, il secondo con una soluzione di nitrato d'argento.
L'isomerismo spaziale (geometrico), altrimenti chiamato isomerismo cis-trans, è caratteristico dei complessi quadrati e ottaedrici (impossibile per quelli tetraedrici). Esempio: isomeria cis-trans di un complesso quadrato

L'isomeria ottica (a specchio) non è essenzialmente diversa dall'isomeria ottica in chimica organica ed è caratteristica dei complessi tetraedrici e ottaedrici (impossibile per quelli quadrati).